TROPIC: Tandem Repeat Locus Prioritization in Cancer
From Spring 2020 to Fall 2021, I engaged in research as an intern in the Snyder Lab at the Genetics Department of Stanford University with Dr. Graham Erwin as my mentor. I co-authored a paper titled “A genome-wide atlas of recurrent repeat expansions in human cancer genomes” that was published in the Nature Journal in December 2022. Below is an abstract of my research.
Background & Motivation
Consecutive repetitive sequences of DNA bases (A,C,T,G) called tandem repeats (eg. …AACAAC…) occur in the human genome. An increase in the number of such repeats is considered a mutation, known as a repeat expansion (eg. …AACAACAACAAC…). Large repeat expansions have already been implicated in severe neurodegenerative disorders. Such large expansions have been suspected but not been extensively investigated in cancer. This work aims to investigate this.
Traditional laboratory techniques (Polymerase Chain Reaction and Southern blot) to identify repeat expansions are time-consuming and tedious since the locations in the genome to investigate are numerous. A much faster recent method uses a computational tool called ExpansionHunter Denovo (EHdn) — from the company Illumina — on fragments of sequenced DNA. EHdn analysis of DNA sequences yields a large number of potential chromosomal locations (loci) at which such repeat expansions can occur. However, there can be errors due to the way repeat expansions are inferred from fragmented DNA sequences. From this vast collection of plausible loci, it is desirable to identify a small ranked set of highly likely loci so that they can be verified in the laboratory. Once such a set of loci is identified, it is also desirable to establish their association with cancer by analyzing genetic and clinical factors.
Objective
My objective was to identify a small subset of repeat expansion loci, which correlate with the occurrences of thirty-eight different cancers, and investigate how these expansions affect gene expression and clinical features, such as tumor stage and grade.
Approach
Towards the objective, I designed and implemented a software pipeline, called TROPIC (Tandem Repeat Locus Prioritization In Cancer), shown in Figure 1. TROPIC analyzed the plausible loci identified by EHdn and cancer patient genetic/clinical data, and consists of four main stages (A, B, C, and D).
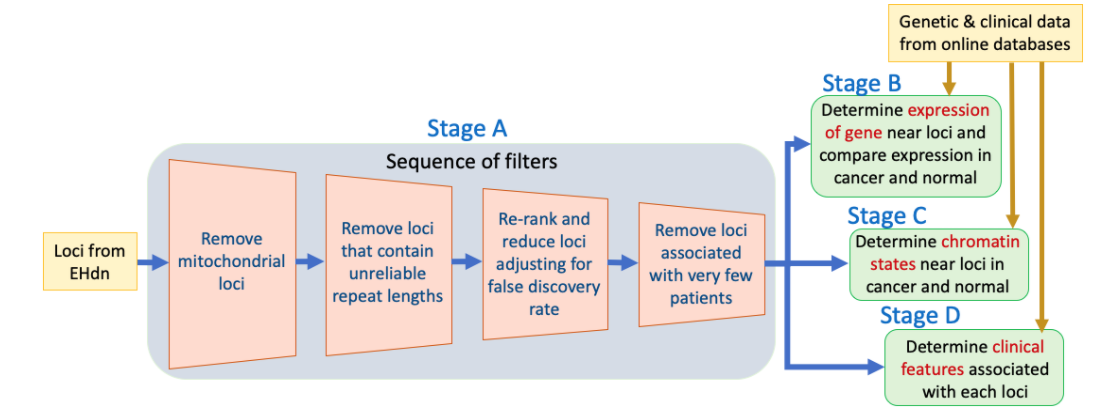
Figure 1. TROPIC stages.
The first stage (Stage A) was developed as a sequence of four software filters to reduce and re-rank the EHdn loci: (1) remove mitochondrial loci, (2) remove loci with patients having unreliable repeat lengths, (3) re-rank and reduce the loci adjusting for false discovery rate, and (4) remove loci that had a very low number of cancer patients with an expansion.
The resulting subset of loci is then analyzed in 3 subsequent stages. Stage B determines expression of the neighboring gene to the loci. Stage C determines the chromatin states in the neighborhood of the loci. Stage D identifies the clinical features, such as sex, tumor stage, and grade of the associated patient. The results of these analyses are visualized across normal and cancer tissues to determine the impact of the loci.
For the above analysis, Dr. Erwin provided me with the plausible loci identified by EHdn, and I gathered cancer patient genetic and clinical data from the International Cancer Genome Consortium (ICGC) Data Portal and organized it into databases. I implemented TROPIC as a combination of programs in Python, R, and Linux Shell. I also created batch automation to complete this analysis for thirty-eight cancers, including breast, ovary, lung, and pancreatic adenocarcinomas, in parallel over a farm of machines.
Results
After analyzing 38 different cancers, I identified 160 loci specific to 7 cancers. For the 31 other cancers, our filtering criteria did not yield any significant loci.